Issues Well-Documented
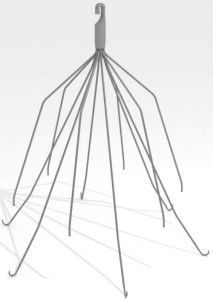
Dodi Froehlich was implanted with an interior vena cava (IVC) filter — a small, spider-shaped medical device designed to stop blood clots from traveling to the heart or lungs — after injuries sustained in a car accident placed her at high risk for clots. Four months later she was in the back of an ambulance, her heart filling with blood and beginning to flat-line.
Doctors performed open-heart surgery on the 45-year old Froehlich and found that a piece of the IVC filter had broken off of the device, traveled to her heart, and pierced it.
Froehlich survived, but other IVC filter recipients haven’t been so lucky.
Gloria Adams, 55, received an IVC filter after she suffered a brain aneurysm. Adams was dead a week after being discharged from the hospital. Like Froehlich, Adam’s heart was punctured by the filter – which was dislodged by a blood clot.
Far from flukes, the IVC filter injuries sustained by Froehlich and Adams are part of a pattern associated with filters made by companies that include C.R. Bard and Cook Medical. More broadly, the potentially dangerous devices shine light on how profit-minded manufacturers conceal product health risks from the public and how loopholes in the medical device approval process can prevent federal regulators from ensuring product safety.
Role of IVC Filters in Pulmonary Embolism Treatment
The interior vena cava is a large-diameter vein that carries deoxygenated blood from the body to the heart. In people who develop blood clots, the vena cava can also carry the clots into vital organs where they have the potential to cause life-threatening injuries.
Anticoagulant drugs are the most common blood clot treatment, but not all patients are good candidates for anticoagulants. For them, an IVC filter —which has a cage-like design that traps blood clots in the vein — may be a safer treatment.
Filters are either retrievable (meant to be removed from the body when blood clot risks subside) or non-retrievable (permanent). Problems with the devices that have emerged in recent years center on the retrievable types.
A Pattern of Harm
A small device placed in a major blood pathway has an inherent risk of dangerous side effects. In addition to complications like bleeding and infection, breakdowns in the devices themselves have been associated with a significant number of adverse events.
Between 2005 and 2010, the FDA received nearly 1,000 IVC filter adverse event reports. The most serious events reported were device migration (328 patient complaints), detachment of device components (146 complaints), IVC perforation (70 complaints), and filter fracture (56 complaints).
They relayed this information in an August 2010 communication which stated that, “These types of events may be related to a retrievable filter remaining in the body for long periods of time, beyond the time when the risk of pulmonary embolism has subsided”.
It was recommended as a precaution that IVC filters be removed from patients as soon as pulmonary embolism protection was no longer needed.
In a follow-up communication released in May 2014, the FDA reiterated this recommendation. They also noted that the entire device could migrate (similar to what happened with Gloria Adams) and that device removal could be difficult. They recommended that physicians evaluate the risks and benefits of device removal for each patient – implying that in some cases removal risks could be unnecessarily hazardous.
These recommendations were based on recently published research and postmarket surveillance studies. One study not referenced by the FDA (published in JAMA in 2013) found that fewer than 10% of IVC filters were successfully removed from patients, while 8% of filter recipients suffered a pulmonary embolism in spite of the device. A companion piece to the JAMA study concluded that, “The IVC filter should capture a blood clot before it reaches the pulmonary circulation and should improve outcomes. This theory, however, has never been validated by empirical studies”.
The companion piece author went on to use the IVC filter to illustrate the “shortcomings of medical device approval in the United States”.
The FDA’s 510(k) Approval Process
Most patients assume that drugs and medical devices prescribed by doctors have been determined to be safe and effective in clinical testing. While this is the case for most drugs and devices it is not true across the board.
As a federal agency with a limited budget, the FDA does not have the resources needed to perform its own clinical testing on each new drug and device that companies hope to market. Manufacturers perform their own clinical testing and the FDA reviews the test data to ensure that it establishes product safety and efficacy.
Product safety is not held to an absolute standard because if new products were expected to produce no harm to any patient under all circumstances, beneficial drugs and devices would almost never make it to market. To promote innovation, the FDA also allows fast-track approval of some new medical devices under what is known as the “510(k)” process. This process allows new medical devices to forego clinical testing if they are said to be “substantially equivalent” to a device already on the market.
The 510(k) process has been heavily criticized by medical experts – with some calling it broken beyond repair. Writing in the prestigious New England Journal of Medicine, Doctors David R. Challoner and William W. Vodra describe how a device that is currently on the market can trace its “substantial equivalence” back to a point where it loses all clinical significance:
Today, we have a system in which a new moderate-risk device can enter the market because it is substantially equivalent to another device that may have been cleared for marketing 2 years ago because its manufacturer showed that it was substantially equivalent to yet another device cleared in 2003, and so on, all the way back to a device that was being marketed when the law was enacted in 1976. But that original device might never have been assessed for safety or effectiveness, nor perhaps would any subsequent ones in the family tree.
About 1/3 of all new medical devices sold in the United States are approved through the 510(k) process. Some of these have caused widespread patient harm, including the infamous DePuy ASR metal hips which had a failure rate approaching 50% and caused severe injuries such as metal poisoning, hip dislocation, and bone fractures.
Bard IVC Recovery
Another 510(k)-approved device is the Bard Recovery IVC filter — the same filter responsible for the death of Gloria Adams and the near-death of Dodi Froehlich. The Recovery IVC is associated with at least 27 deaths and more than 300 injuries and has a failure rate of 16 percent.
Bard was not successful in its first attempt at 510(k) approval for the Recovery, submitted in 2002. In response, Bard hired regulatory specialist Kay Fuller to help with the approval process. Fuller expressed concerns over the Recovery’s safety. Specifically, she said that Bard withheld important safety evidence from her and that the evidence she did see was troubling. When Fuller voiced her concerns, she said that Bard threatened to dismiss her from the team. Despite her refusal to sign an FDA application for the device, Fuller’s signature appeared on the document — an apparent forgery.
Fuller expressed her concerns to the FDA but the agency has yet to formally address them. They did, however, send a warning letter to Bard in July 2015 reprimanding the manufacturer for failing to properly establish and maintain procedures for reviewing and evaluating complaints relating to its G2, G2X and Eclipse IVC filters.
The Bard G2 IVC filter was also approved under the 510(k) approval process after it was deemed to be substantially equivalent to the problematic Recovery IVC filter. The G2 filter has been shown to have a 12% failure rate.
Other IVC filters approved through the 510(k) process include the Cook Günther Tulip Vena Cava Filter and the Cook® Celect ®Vena Cava Filter. These Cook IVCs were found in a 2012 study to have a 100% vena cava perforation rate and a 40% device tilt rate.
Bard May Have Withheld Risks From Public
Evidence suggests that Bard knew about dangers with the Recovery filter but failed to share this information with the public.
The evidence comes from a report commissioned by Bard and obtained by NBC News. It shows that the device maker knew perhaps as early as 2004 that the Recovery, compared to all of its competitors, was more likely to fracture, migrate, and cause patient death. But Bard failed to offer a recall. It sold the device for 3 more years, over which time it was implanted in 34,000 patients. Then Bard modified, renamed, and remarketed the device as the G2 – which is implanted in an estimated 62,000 patients.
Victims File Lawsuits Against Filter Makers
Since suffering a punctured heart that almost killed her, Dodi Froehlich has filed a lawsuit against C.R. Bard, Inc. She is joined by dozens of other device recipients who allege to also have suffered injuries caused by their Bard IVC filters.
A multidistrict litigation (MDL) for all Bard IVC filter claims is currently consolidated in an Arizona U.S. District Court. As of September 15, 2015, the MDL contains 50 separate injury claims.
Cook Medical is also dealing with an IVC MDL consolidated in Southern Indiana U.S. District Court. The Cook MDL currently has 120 claims. Similar to the Bard IVC MDL, the Cook MDL is related to the alleged ability of Cook IVC filters to cause injuries when they fracture, migrate, tilt, and perforate.
Sources
- http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm221707.htm
- http://www.nbcnews.com/health/health-news/did-blood-clot-filter-used-thousands-americans-have-fatal-flaw-n384536
- http://archinte.jamanetwork.com/article.aspx?articleid=1669107
- http://www.medscape.com/viewarticle/744331_2
- http://www.nejm.org/doi/full/10.1056/NEJMp1109150?viewType=Print&
